überblick
Details zur Erkrankung
Eine Vielzahl genetischer Erkrankungen führt zur Ausbildung einer spinalen Deformität, welche meist im Kindes- oder Jugendlichenalter auftritt. Die spinalen Deformitäten bei diesen Menschen kommen häufiger vor als die idiopathische Skoliose in der Gesamtbevölkerung, wobei die Inzidenz von Grunderkrankung zu Grunderkrankung variiert. Diese Deformitäten sind Skoliosen, welche oft jedoch eine besonders ausgeprägte lordotische oder kyphotische Komponente haben. Skoliosen also welche im Rahmen genetischer Syndrome auftreten werden syndromale Skoliose genannt. Die häufigsten genetischen Syndrome, welche mit spinalen Deformitäten einhergehen sind: Down Syndrom, Ehlers-Danlos Erkrankungen, Friedreich Ataxie, Marfan Syndrom, Neurofibromatose, Osteogenesis imperfecta, Prader Willi Syndrom und Rett Syndrom. Da sich die klinischen Charakteristika der spinalen Deformitäten von genetischem Syndrom zu genetischem Syndrom variieren, wird im Folgenden auf jedes Einzelne eingegangen.
Down Syndrom
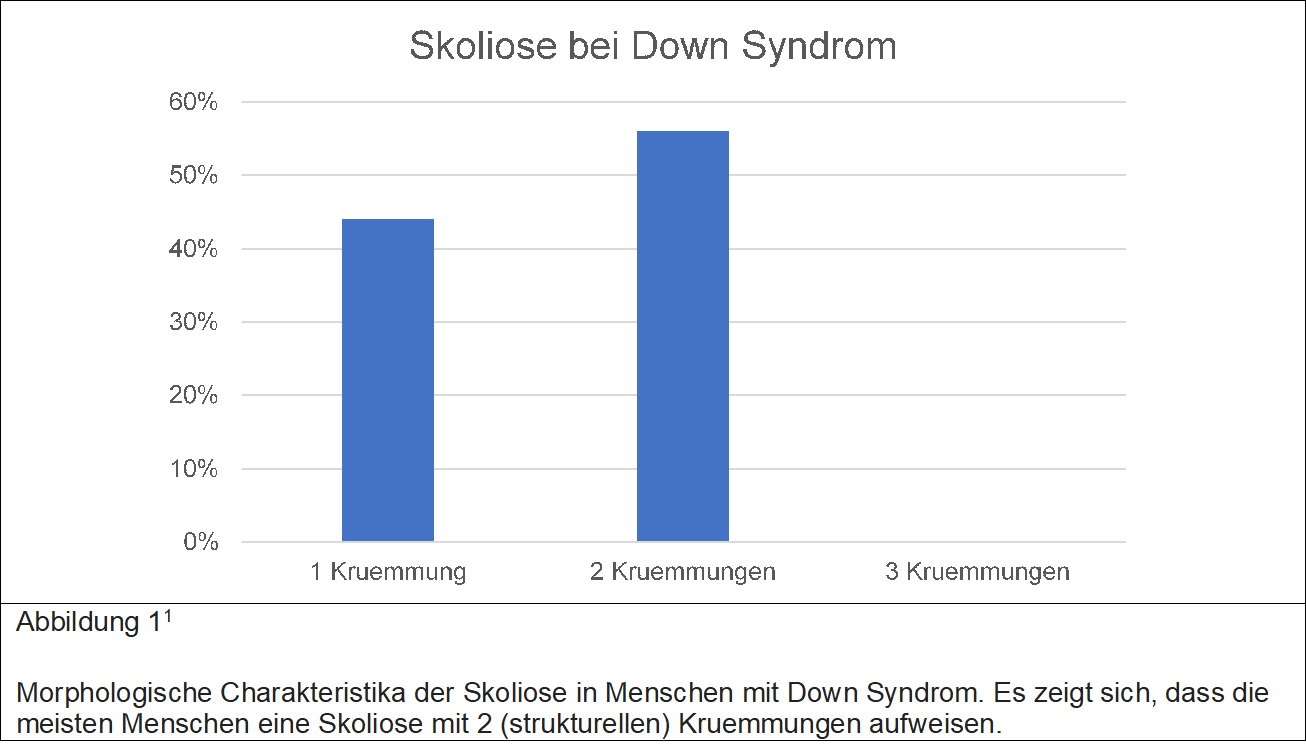
Eine genetische Erkrankung, welche etwa 1 pro 700 Menschen betrifft. Betroffene Menschen leiden mitunter unter Herzerkrankungen, Skoliose, Instabilität am Übergang Kopf zu Halswirbelsäule, Gelenksinstabilität oder auch entzündliche Darmerkrankungen. Die Tabelle 1 gibt eine kurze Uebersicht ueber das Auftreten einer Skoliose beim Down Soyndrom. Ist eine Skoliose vorhanden, so wird ab 35° Krümmungswinkel (Cobb Winkel) eine Korsetttherapie empfohlen, obwohl, diese in den meisten Fällen nicht erfolgreich ist. Die Indikation zur operativen Therapie hängt von mehrerlei Faktoren ab, unter anderem Skelettalter, und Cobb Winkel. Die Abbildung 1 zeigt die morphologischen Charakteristika skoliotischer Veränderungen bei Menschen mit Down Syndrom. Es ist von besonderer Relevanz zu erwähnen, dass sein Teil der Menschen mit Down Syndrom auch an Gelenksinstabilität am kraniocervicalen Übergang leidet. Ein grosser Teil der Menschen mit Down Syndrom muss sich zu einem Zeitpunkt einer Herzoperation unterziehen. Diese beiden Faktoren sind wesentlich zu berücksichtigen Falls über die operative Korrektur einer Skoliose diskutiert wird.

Ehlers-Danlos Syndrom
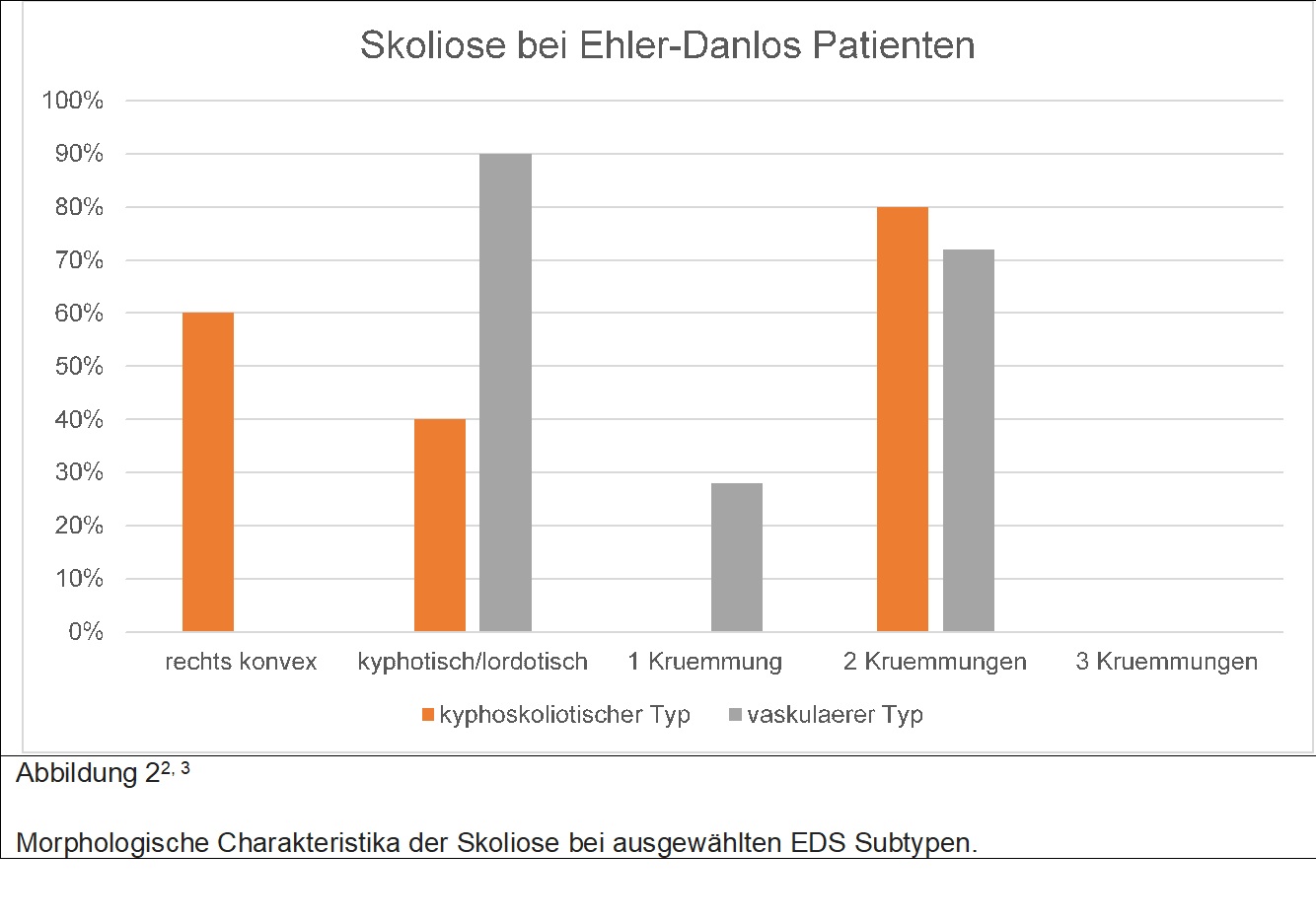
Ist eine Gruppe von 13 verschiedenen genetischen Erkrankungen des Bindegewebes, welches sich zum Teil sehr unterschiedlich manifestieren. Während beispielsweise Patienten mit dem vaskulären Krankheitstyp in erster Linie durch Gefässerkrankungen auffallen (sogenannte Dissektionen und Aneurysmen an Gefässen im ganzen Körper), so manifestieren sich andere Subtypen wie der Hypermobile in Form von Gelenksinstabilität und Luxationen. Vielen EDS Typen ist ein chronisches Schmerzsyndrom gemeinsam. Skoliose kann auch bei EDS Patienten auftreten, jedoch ist das Zusammenspiel mit der Grunderkrankung etwas komplexer. Die Tabelle 2 gibt eine Übersicht welche EDS Subtypen häufig zu Skoliose führen.
Wie sich aus der Tabelle 2 ergibt, so gibt es EDS Subtypen, welche oft mit milderen Skoliosen einhergehen (vaskulärer und hypermobiler Typ), und auch solche, welche mit ausgeprägteren Skoliosen einhergehen (kyphoskoliotischer und muskulokontrakturaler Typ). Patienten mit vaskulärer oder hypermobiler EDS haben oft Skoliosen, welche mehr einer idiopathischen Skoliose ähnlich sind, also weniger ausgeprägter Cobb Winkel, meist keine wesentliche Kyphose oder Lordose. Die Behandlung dieser Patienten ähnelt mehr der Patienten mit idiopathischer Skoliose. Hingegen ist die Behandlung der schwerer verlaufenden Subtypen dahingehen anders, als dass Korsetttherapie oft nicht erfolgreich ist und eine operative Korrektur notwendig wird. Bei mehreren EDS Subtypen, vor allem der vaskulären und kyphoskoliotischen Form ist sehr wichtig zu wissen, dass die Gefasse dieser Patienten sehr fragil sind, was einen Einfluss auf das Komplikationsprofil einer Operation haben kann.
Friedreich Ataxie
Ist eine erblich bedingte neurodegenerative Erkrankung, welche zwischen 1-2 Menschen pro 50000 Einwohner betrifft. Die ersten Krankheitssymptome treten meist in der Pubertät auf und bestehen aus Gangstörungen und Muskelschwäche. Weitere Symptome sind Bewegungsstörungen, Herzerkrankungen, Sprach-, Seh- und Hörstörungen. Obwohl der Krankheitsverlauf zwischen Patienten unterschiedlich sein kann, wird nach mehreren Jahren schlussendlich ein Rollstuhl benötigt. Eine grosse Anzahl der Patienten entwickeln eine Skoliose, von denen jedoch viele (anders als bei anderen syndromalen Skoliosen) auf eine Korsetttherapie ansprechen. Schlussendlich benötigen im Schnitt 19% eine operative Korrektur. Details siehe Tabelle 3.
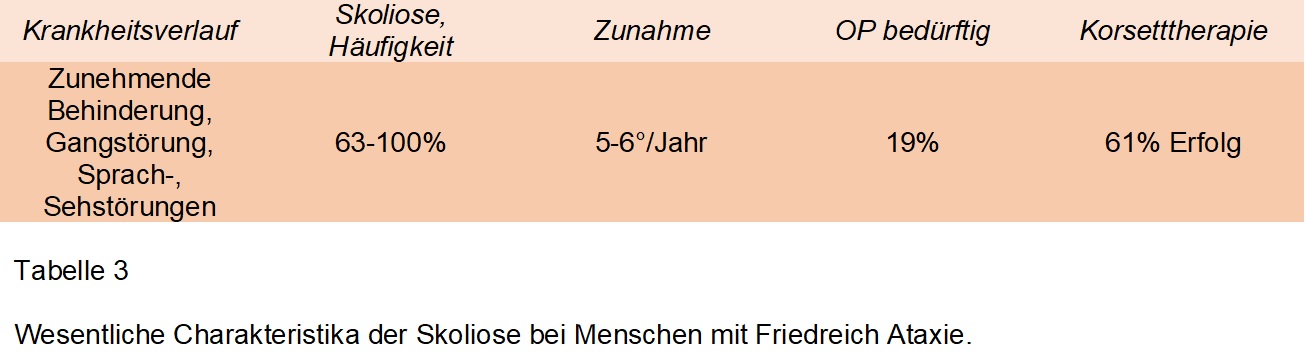
Für eine Zunahme der Deformität sprechen Doppelkurven, frühes Erkrankungsalter mit, jedoch nicht Schwere der Friedreich Ataxie. Korsetttherapie war in bis zu 61% der Patienten erfolgreich, jedoch bedurften schlussendlich 19% einer operativen Korrektur. Die Abbildung 3 zeigt die morphologischen Charakteristika der Skoliose bei Menschen mit Friedreich Ataxie.
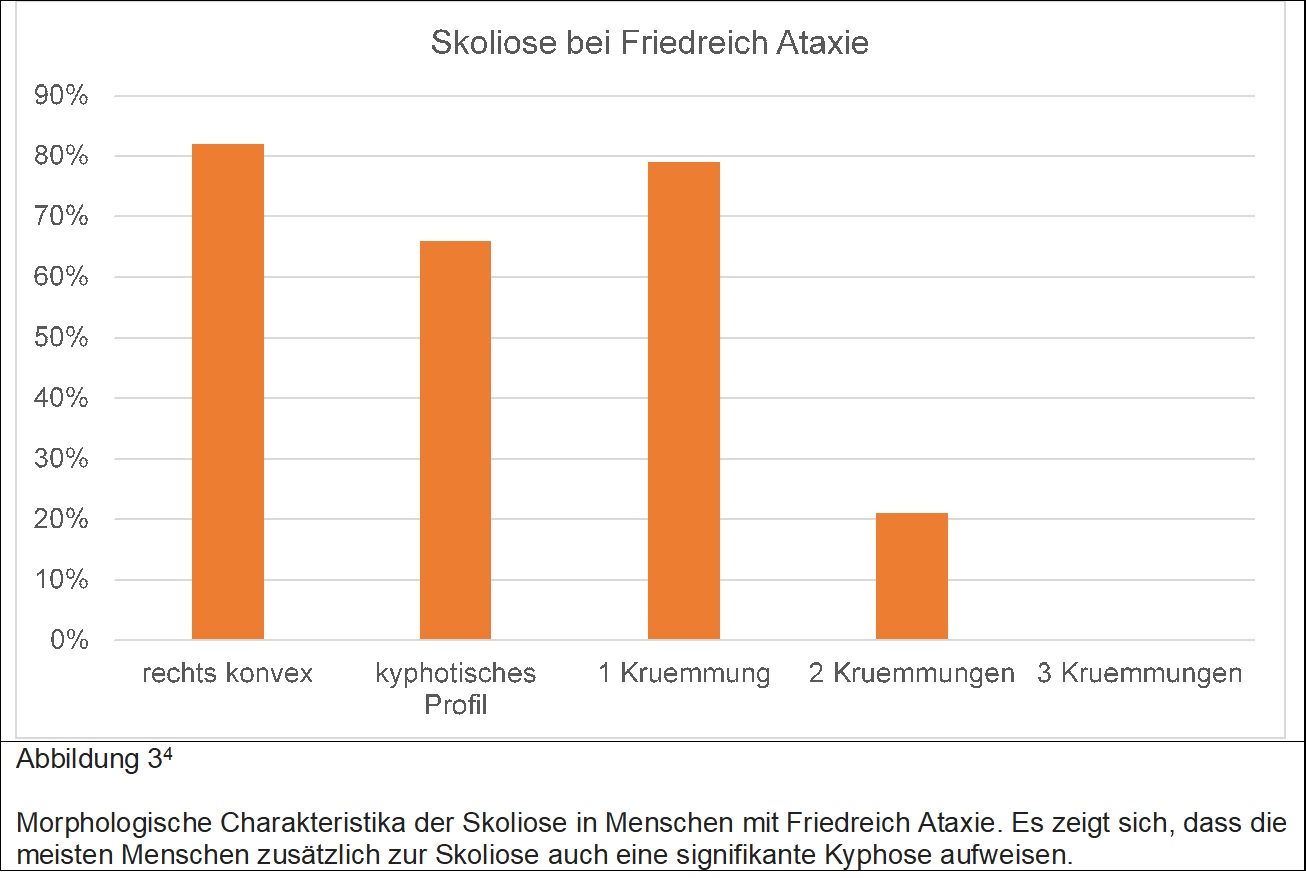
Koresetttherapie wird begonnen ab einem Cobb Winkel von 15° vor dem Wachstumsschub der Pubertät und 20° danach. Wesentliche morphologische Unterschiede zur idiopathischen Skoliose sind das häufige Vorhandensein einer thorakalen Kyphose sowie einer links konvexen Krümmung.Operative Indikationen sind: eine Zunahme >5°/ Jahr nach dem pubertären Wachstumsschub, eine maximale Krümmung von mehr als 40°. Wenn Patienten eine signifikante koronare oder sagittal Imbalance haben, ist die Operation gegebenenfalls bei weniger Krümmung notwendig. Anders als bei der idiopathischen Skoliose wäre bei vielen Patienten eine Ausdehnung der Versteifung bis zum Becken notwendig, was jedoch von den meisten Zentren nur bei rollstuhlpfllichtigen Patienten durchgeführt wird.
Marfan Syndrom
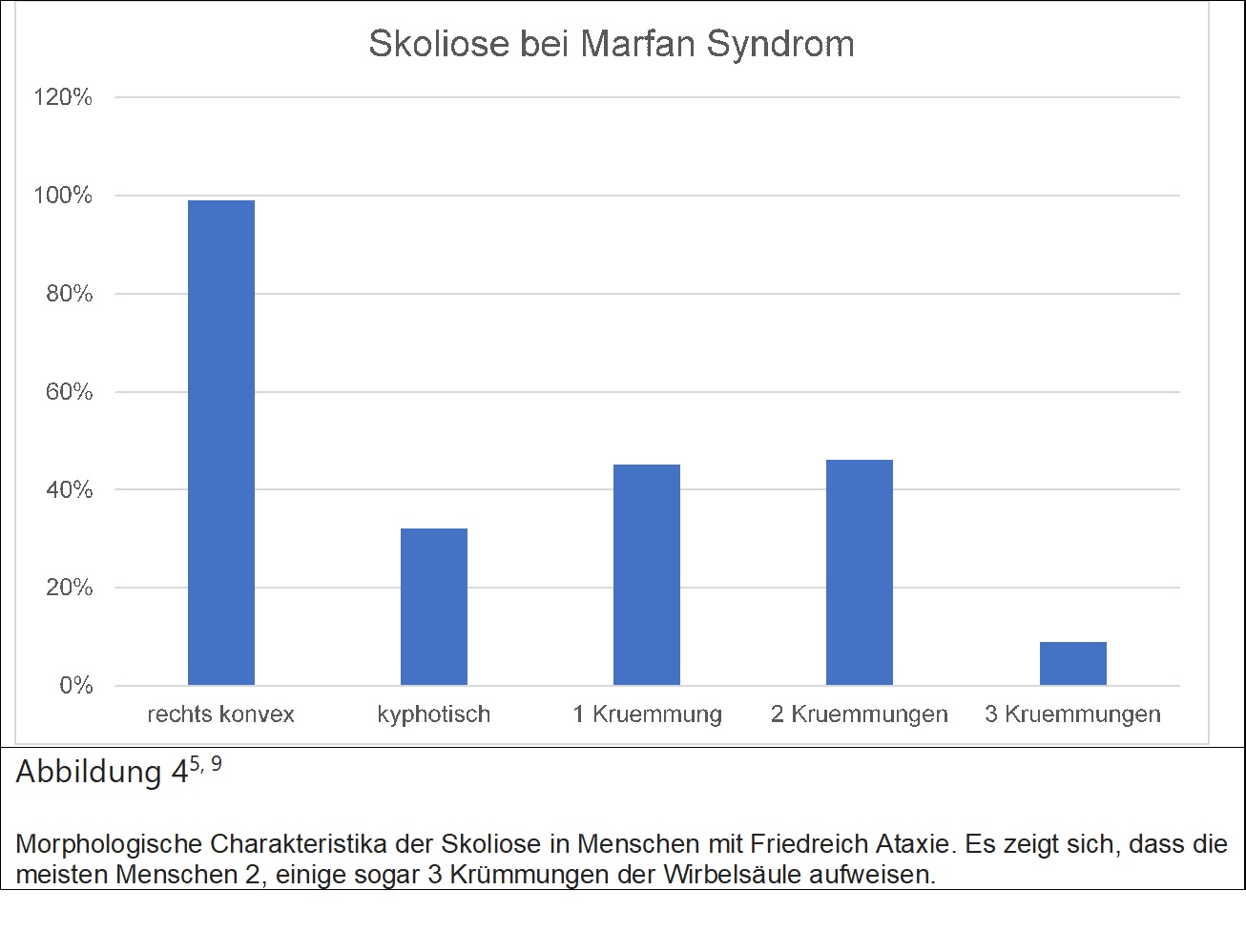
Eine erbliche Bindegewebserkrankung, welche etwa 2-3 / 100000 Menschen betrifft und sich folgendermassen manifestieren kann: häufig betroffen sind das Herz-Keislaufsystem, wobei es hier zu Rissen in den Gefässen kommen kann (Dissektionen) oder auch Ausstülpungen derselben (Aneurysmen). Weiteres kommt es oft zu Schädigungen der Herzklappen, Störungen der Augenlinsen und Skoliose. Die wichtigsten Charakteristika zu Marfan Syndrom und der damit oft verbundenen Skoliose finden sich in Tabelle 4.

Häufiger als bei idiopathischer Skoliose tritt die Deformität bei Marfan Patienten oft schon im Kindesalter auf. Bei einem Cobb Winkel zwischen 20° und 40° wird ein Korsett angewendet, was jedoch nur in 17% erfolgreich ist. Eine operative Therapie wird ab 40° indiziert. Besonders behandlungsrelevant ist das häufige Vorkommen lumbosacraler Übergangswirbel, das Vorhandensein von Duralektasien sowie ausgedünnter Pedikeln.
Neurofibromatose
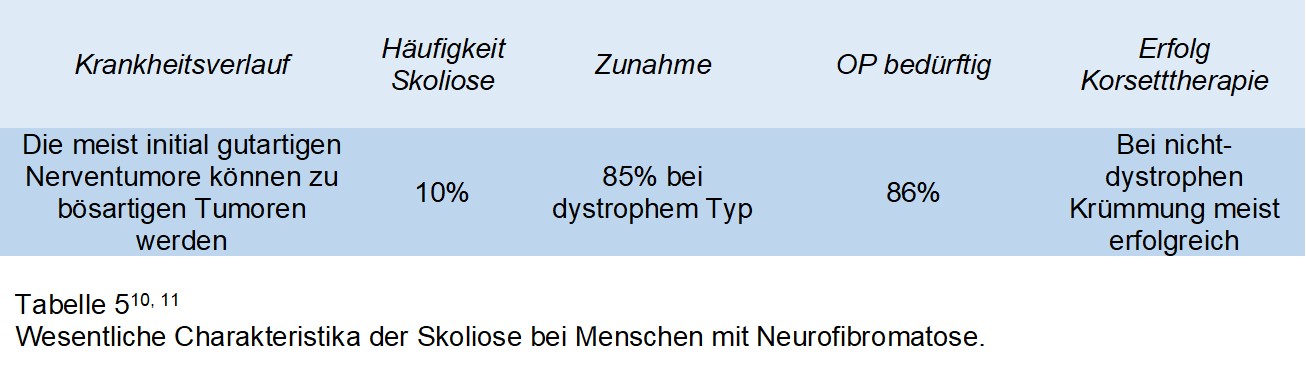
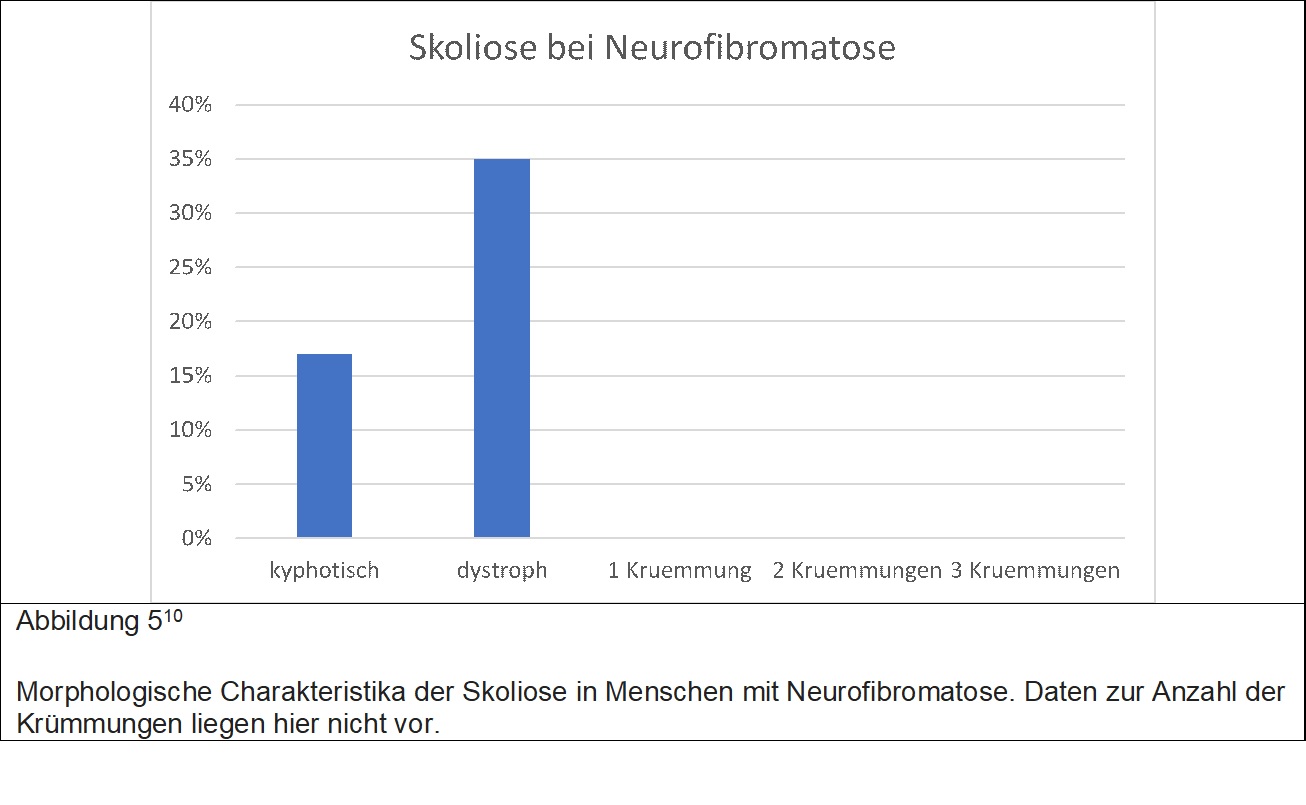
Eine genetische Erkrankung welche etwa 1 / 3000 Menschen betrifft und in 2 Gruppen vorkommt. Bei dem Typ 1, der von Recklinghausen Erkrankung, leiden die Betroffenen an einer Vielzahl von meist gutartigen Nervenscheidentumoren, welche nahezu am ganzen Körper auftreten können, sowie in einigen Fällen epileptische Anfälle, kognitive und Verhaltensauffälligkeiten wie auch Skoliose. Bei der Neurofibromatose vom Typ 2 leiden die betroffenen an gutartigen Tumoren der Hör- und Gleichgewichtsnerven und in einigen Fällen auch an Tumoren der Hirnhäute und des Rückenmarkes. Für den Chirurgen wichtig zu wissen ist auch die Tatsache das eine Teil er NF Patienten an Ausstülpungen der Rückenmarkshaut leiden, welche zwar per se keine Symptome verursacht, jedoch zu operative Komplikationen führen kann. Die Skoliose kann morphologisch der AIS ähnlich sein, jedoch kann es in einigen Menschen auch zur Entwicklung von sog. dystrophen skoliotischen Krümmungen kommen, welche einer mehr angulareren Deformität entsprechen. Bei Menschen mit NF1 kommt es häufig zu knöchernen Veränderungen an der Wirbelsäule, sogenannte dysplastische Veränderungen. Sind solche Veränderungen vorhanden, dann ist das Risiko für eine Zunahme der Skoliose besonders hoch. Patienten mit dystropher Krümmung erleben in 85% eine Zunahme der Skoliose. Ist eine operative Korrektur notwendig, ist es besonders wichtig vor der Operation eine MRI Untersuchung der gesamten Wirbelsäule durchzuführen, da Menschen mit NF1 in etwa einem Drittel gutartige Tumore innerhalb des Wirbelkanals aufweisen.Patienten mit einer nicht-dystrophen Krümmung werden ähnlich behandelt wie Patienten mit idiopathischer Skoliose. Bis zu einem Krümmungswinkel von 25° erfolgt weder Korsettbehandlung noch OP, lediglich wird der Patient alle 6 Monate mit Roentgen kontrolliert (in der pubertären Wachstumsphase). Patienten mit einer Krümmung zwischen 20° und 40° sollten ein Korsett tragen und über 40° sollte eine operative Korrektur durchgeführt werden. Patienten mit einer dystrophen Krümmung, also eine scharf angulaere Krümmung, ist eine wesentlich andere Therapie notwendig, da diese Krümmungen ohne Behandlung deutlich Zunahmen während der Pubertät und nicht gut auf Korsetttherapie ansprechen. Entsprechend ist eine frühzeitige operative Korrektur notwendig. Bei diesen Patienten kann eine engmaschige Kontrolle bei Krümmungen bis 20° vorgenommen werden. Ist die Krümmung höher so ist eine operative Korrektur indiziert. Es ist bei der Verlaufsbeobachtung anzumerken, dass Patienten mit zunächst einer nicht dystrophen Krümmung im Verlauf eine dystrophe Krümmung entwickeln. Tabelle 5 und Abbildung 5 zeigen eine Übersicht über die wesentlichen Charakteristika der Skoliose bei Menschen mit Neurofibromatose.
Osteogenesis imperfecta
Osteogenesis imperfecta ist eine genetische Erkrankung, bei der eine Komponente des Bindegewebes, das Kollagen vom Typ I, in der Bildung gestört ist. Die Erkrankung kommt mit einer Häufigkeit von 1/10000-20000 Menschen vor und wird in 4 Gruppen eingeteilt. Die möglichen Symptome der Erkrankung sind sehr verschieden, jedoch wesentlich geprägt davon um welchen Subtype es sich handelt. Tabelle 6 zeigt die Klassifizierung.

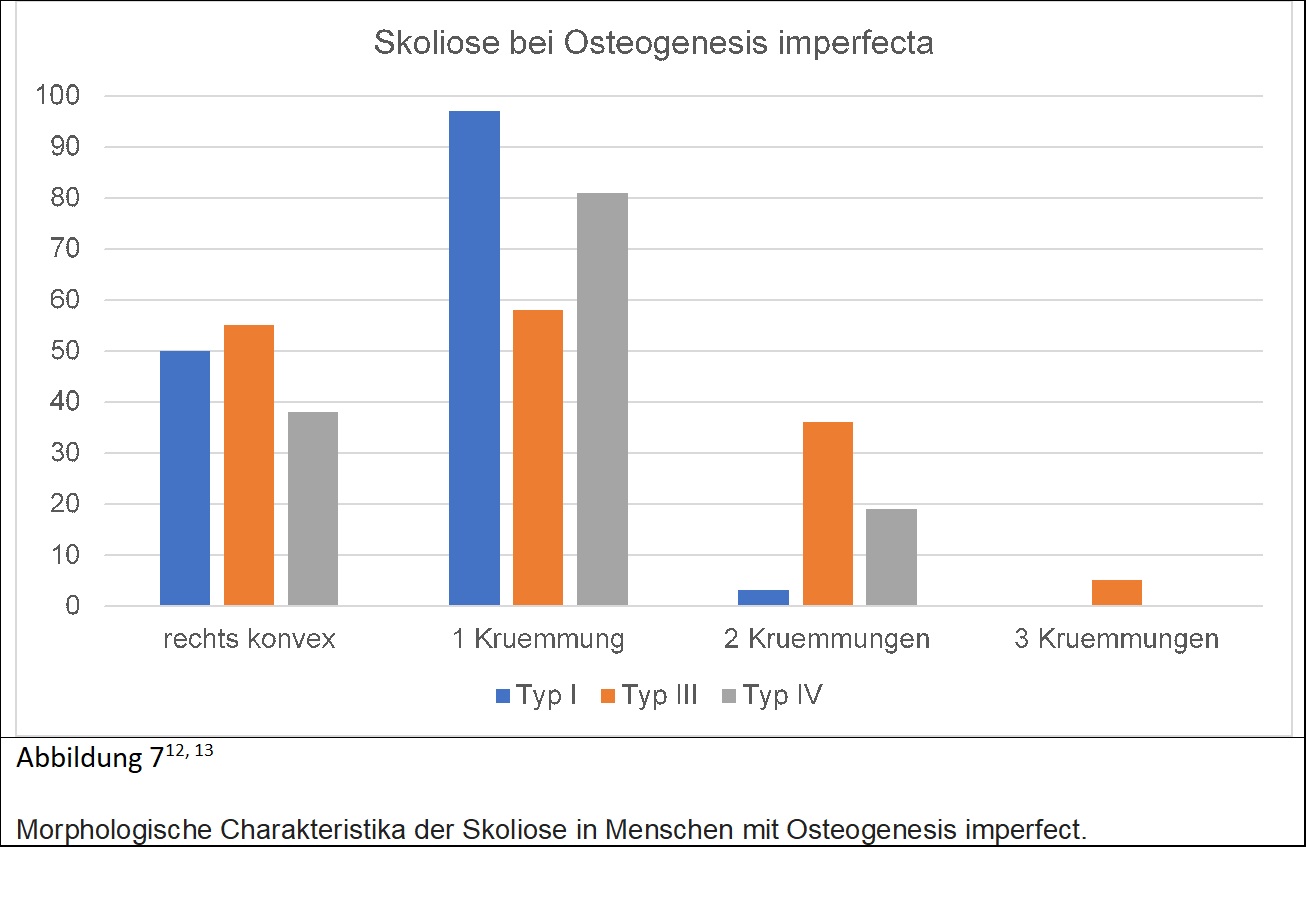
Wie der Klassifizierung zu entnehmen ist, kann die Gabe eines Medikamentes, namentlich Diphosphonat, vor dem Alter von 6 Jahren die Progression der Skoliose deutlich verlangsamen. Abgesehen vom Typ der der Osteogenesis imperfecta sind noch weitere Faktoren vergesellschaftet mit einer deutlicheren Zunahme der Skoliose, nämlich niedrige Knochendichte sowie das Vorhandensein von 6 oder mehr biconcav deformierten Wirbelkörpern. Die Behandlung ist schwierig auf Grund der schlechten Knochenqualität. Anders als bei der idiopathischen Skoliose tendierte Skoliose bei Osteogenesis imperfecta auch im Erwachsenenalter deutlich zuzunehmen, vor allem in den Subgruppen III und IV.Patienten mit einem Cobb Winkel unter 30° werden jährlich kontrolliert, zwischen 30° und 50° häufiger. Eine Krümmung von mehr als 50° wird als Indikation zur Operation gesehen. Ab einem Cobb Winkel von 60° tritt eine merkliche Reduktion der Lungenfunktion um 50% ein, was die Lebensqualität merklich beeinträchtigt. Ein weiteres wichtiges Kriterium ist die Flexibilität der Krümmung. Ab einer Krümmung von 90° wird meist vor der OP Traktion angewendet. Eine präoperativ begonnene Bisphosphonat Therapie hat günstigen Effekt auf den Knochen. Weiters anzumerken ist, dass es bei Patienten mit OI häufig zu kyphotischen Deformitäten am kranicervialen Übergang kommen kann, was vor einer Operation in Bauchlage unter Umständen abzuklären ist. Die Abbildung 7 zeigt Details zur Form der Skoliose bei Menschen mit Osteogenesis imperfecta.
Operative Behandlung
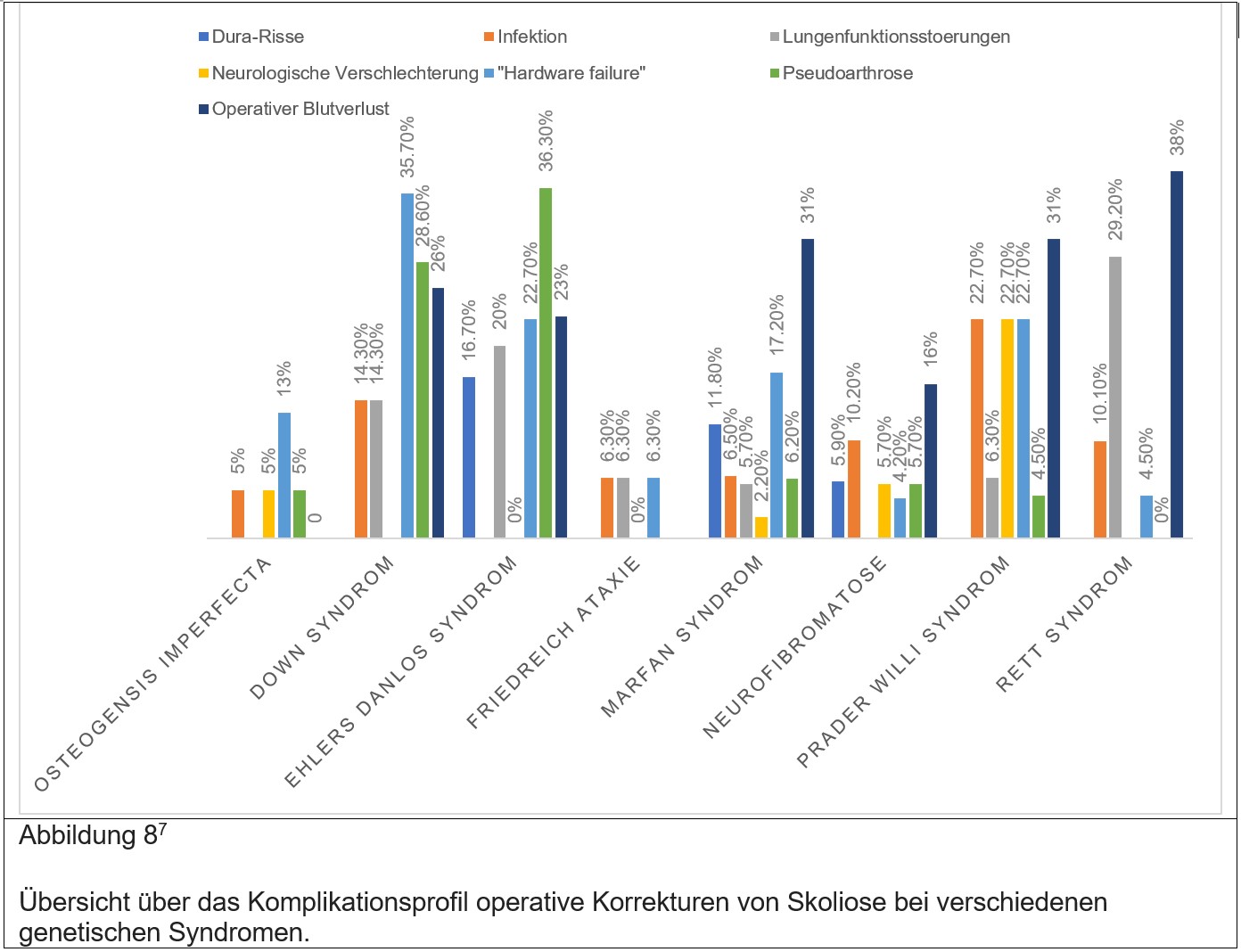
Wie bereits in den Unterkapiteln zu den einzelnen Syndromen beschrieben, so unterscheidet sich die Behandlung syndromaler Skoliosen oft von der der idiopathischen Skoliose. Ein weiterer Unterschied liegt darin, dass Menschen mit einem genetischen Syndrom oft anfälliger sind operative und perioperative Komplikationen zu erleiden als Menschen ohne genetischem Syndrom, welche an einer einfachen idiopathischen Skoliose leiden. Weiters zeigen sich skoliotische Krümmungen in Menschen mit genetischem Syndrom oft morphologisch unterschiedlich (z.B. Anzahl der Krümmungen). Schlussendlich ist bei den meisten syndromalen Skoliosen das Risiko eine operative Komplikation zu erleiden auf Grund des eben Erwähnten höher. Die Abbildung 8 und 9 zeigen eine Übersicht operativer Komplikationen.
Weiterführende Literatur
Friedreich Ataxie Parent Support
Referenzen
1. Milbrandt TA, Johnston CE, 2nd. Down syndrome and scoliosis: a review of a 50-year experience at one institution. Spine (Phila Pa 1976). 2005;30(18): 2051-2055. https://doi.org/10.1097/01.brs.0000179100.54453.72.
2. Akpinar S, Gogus A, Talu U, Hamzaoglu A, Dikici F. Surgical management of the spinal deformity in Ehlers-Danlos syndrome type VI. Eur Spine J. 2003;12(2): 135-140. https://doi.org/10.1007/s00586-002-0507-6.
3. Jasiewicz B, Potaczek T, Tesiorowski M, Lokas K. Spine deformities in patients with Ehlers-Danlos syndrome, type IV – late results of surgical treatment. Scoliosis. 2010;5: 26. https://doi.org/10.1186/1748-7161-5-26.
4. Simon AL, Meyblum J, Roche B, et al. Scoliosis in Patients With Friedreich Ataxia: Results of a Consecutive Prospective Series. Spine Deform. 2019;7(5): 812-821. https://doi.org/10.1016/j.jspd.2019.02.005.
5. Garreau de Loubresse C, Mullins MM, Moura B, Marmorat JL, Piriou P, Judet T. Spinal and pelvic parameters in Marfan’s syndrome and their relevance to surgical planning. J Bone Joint Surg Br. 2006;88(4): 515-519. https://doi.org/10.1302/0301-620X.88B4.17034.
6. Peterson LE, Nachemson AL. Prediction of progression of the curve in girls who have adolescent idiopathic scoliosis of moderate severity. Logistic regression analysis based on data from The Brace Study of the Scoliosis Research Society. J Bone Joint Surg Am. 1995;77(6): 823-827. https://doi.org/10.2106/00004623-199506000-00002.
7. Levy BJ, Schulz JF, Fornari ED, Wollowick AL. Complications associated with surgical repair of syndromic scoliosis. Scoliosis. 2015;10: 14. https://doi.org/10.1186/s13013-015-0035-x.
8. Sponseller PD, Bhimani M, Solacoff D, Dormans JP. Results of brace treatment of scoliosis in Marfan syndrome. Spine (Phila Pa 1976). 2000;25(18): 2350-2354. https://doi.org/10.1097/00007632-200009150-00013.
9. Sponseller PD, Hobbs W, Riley LH, 3rd, Pyeritz RE. The thoracolumbar spine in Marfan syndrome. J Bone Joint Surg Am. 1995;77(6): 867-876. https://doi.org/10.2106/00004623-199506000-00007.
10. Akbarnia BA, Gabriel KR, Beckman E, Chalk D. Prevalence of scoliosis in neurofibromatosis. Spine (Phila Pa 1976). 1992;17(8 Suppl): S244-248. https://doi.org/10.1097/00007632-199208001-00005.
11. Crawford AH. Pitfalls of spinal deformities associated with neurofibromatosis in children. Clin Orthop Relat Res. 1989(245): 29-42.
12. Anissipour AK, Hammerberg KW, Caudill A, et al. Behavior of scoliosis during growth in children with osteogenesis imperfecta. J Bone Joint Surg Am. 2014;96(3): 237-243. https://doi.org/10.2106/JBJS.L.01596.
13. Wallace MJ, Kruse RW, Shah SA. The Spine in Patients With Osteogenesis Imperfecta. J Am Acad Orthop Surg. 2017;25(2): 100-109. https://doi.org/10.5435/JAAOS-D-15-00169.